21July, 2023
Adrenal disorders and fertility
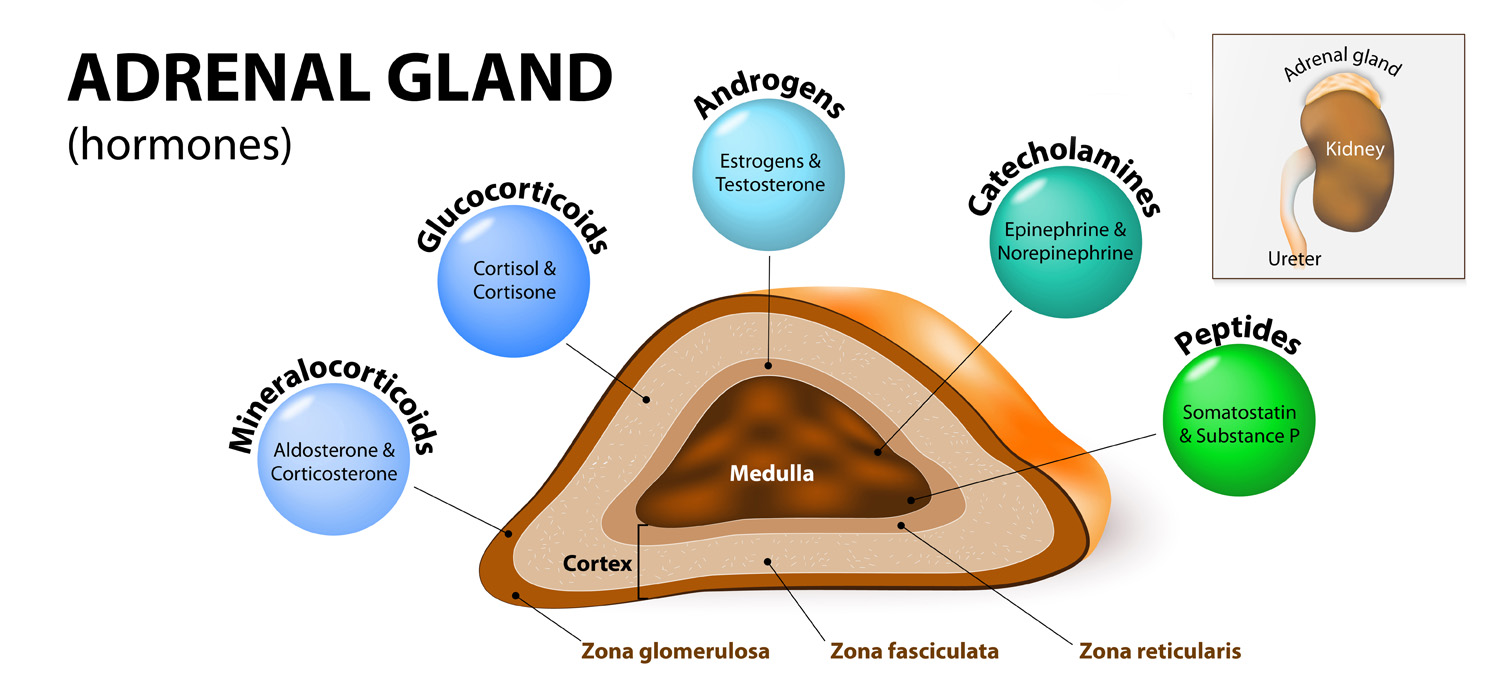
The adrenal medulla produces epinephrine under the control of the sympathetic nervous system. The adrenal cortex has:
- Zona Glomerulosa- secretes Aldosterone- under control of Renin-Angiotensin system.
- Zona Fasciculata- Secretes Cortisol-under ACTH control.
- Zona Reticularis-Under ACTH control
Cushing’s syndrome
Occurs due to excessive exposure to Cortisol. The source of steroids may be iatrogenic or endogenous. The latter cause is further grouped as ACTH-dependent and ACTH-independent.
The most common cause of this syndrome is ACTH-producing pituitary tumors (Cushing Disease). Other causes are carcinoma, bronchial carcinoids, and neuroendocrine tumors.
Features include central obesity (moon facies and buffalo hump), hirsutism, proximal muscle weakness, thin skin with easy bruisability and formation of striae. Young women with PCOS (hyperandrogenism+anoulation+PCOM morphology) with dermatological manifestation must be considered in differential diagnosis.
Diagnosis
Diagnosis is difficult in the initial stages.
Excessive cortisol production with loss of diurnal variation. 24-hour urinary free cortisol, serum and salivary cortisol measurements reflect the excess steroids. Dexamethasone suppression fails to reduce the cortisol levels in blood.
ACTH is increased in ACTH dependent Cushing’s. MRI with Gadolinium contrast can be done to define the pituitary tumor. CT scan of adrenal glad is done to identify local causes if ACTH is found to be low.
Treatment
Surgery is the choice in Cushing’s Syndrome. Transsphenoidal pituitary adenectomy is done in those with Cushing’s disease and if unsuccessful may require radiotherapy or bilateral adrenalectomy.
Bilateral or unilateral adenectomy is done in non-ACTH dependent Cushing’s syndrome and treatment resistant Cushing’s disease.
Ketoconazole, mifepristone and Cabergoline have been tried for medical management.
Congenital Adrenal Hyperplasia
Group of autosomal recessive metabolic disorders characterized by abnormal metabolism in cholesterol pathways resulting I low levels of cortisol. The most common is 21-hydroxylase deficiency, with mutation of the gene occurring on Chromosome 6.
Depending on severity of the disease and age of presentation, CAH is classified as classical virilizing CAH, simple virilizing CAH and non-classical CAH or late-onset CAH. (NC-CAH)
Classical virilizing CAH presents at birth with ambiguous genitalia in girls and may be associated with salt losing condition with dyselectrolytemia and can be fatal then. Routine newborn screening with Guthrie cards can identify the condition so that treatment can be started immediately, reducing morbidity and mortality.
Simple virilizing CAH presents precocious puberty, clitoromegaly, virilization and early skeletal maturity.
NC-CAH presents in peri-pubertal girls with primary amenorrhea or oligomenorrhoea, hirsutism, clitoromegaly or acne. Scan may show polycystic ovarian morphology (PCOM) and these cases must be differentiated for PCOS. Serum cortisol < 50ng/ml, 17-OHP > 10,000, high urinary ketosteroids offer a definitive diagnosis.
Management
Newborns with ambiguous genitalia merit karyotyping, abdomen ultrasound for uterus/ovaries and CAH screen with special attention to the salt-wasting type of CAH. Steroid replacement and counselling with family support are the mainstays of treatment in CAH.
For more info, Visit : www.medlineacademics.com